It really represents a very complex mechanism -- the cough reflex arc.
There are cough receptors from the upper airway leading all the way down
to the terminal bronchioles, not including, however, the terminal
bronchioles and the alveoli. There are cough receptors in the nose and
there are cough receptors in the ear canal. So some of you may have had
patients who began coughing when you began doing otoscopic examinations
-- and that's why, because you began stimulating their cough receptors.
These afferent impulses travel via the vagus nerve to the medullary
cough center, and from there a very complex but integrated mechanism
occurs traveling back through efferent vagus nerves that go to the
larynx, the chest muscles, the diaphragm, abdominal wall, pelvic floor,
and the bronchii themselves. And a very complex and very integrated
motor response that leads ultimately to what we see, finally, as the
cough.
http://www.medscape.org/viewarticle/430253
Sunday, January 8, 2012
Dexamethasone suppression test
How the test is performed:
There are two different types of dexamethasone suppression tests: the low-dose test and the high-dose test. Each type can either be done in an overnight or standard (3-day) way.
For the low-dose overnight method, 1 mg of dexamethasone is given at 11 p.m., and the blood is drawn at 8 a.m. for a cortisol measurement (see venipuncture).
In the standard low-dose method, urine is collected 3 days (stored in 24-hour collection containers) for measurement of cortisol. On day 2, a low dose (0.5 mg) of dexamethasone is given by mouth every 6 hours for 48 hours.
For the high-dose overnight method, a baseline cortisol is measured on the morning of the test, then 8 mg of dexamethasone is given at 11 p.m. Blood is drawn at 8 a.m. for a cortisol measurement. For the standard high-dose test, urine is collected over 3 days (stored in 24-hour collection containers) for measurement of cortisol. On day 2, a high dose (2 mg) of dexamethasone is given by mouth every 6 hours for 48 hours.
What abnormal results mean:
If there is not a normal response on the low-dose test, abnormal secretion of cortisol is likely (Cushing's Syndrome). This could be a result of a cortisol-producing adrenal tumor, a pituitary tumor that produces ACTH, or a tumor in the body that inappropriately produces ACTH. The high-dose test can help distinguish a pituitary cause (Cushing's Disease) from the others.
Cushing's syndrome caused by adrenal tumor
There are two different types of dexamethasone suppression tests: the low-dose test and the high-dose test. Each type can either be done in an overnight or standard (3-day) way.
For the low-dose overnight method, 1 mg of dexamethasone is given at 11 p.m., and the blood is drawn at 8 a.m. for a cortisol measurement (see venipuncture).
In the standard low-dose method, urine is collected 3 days (stored in 24-hour collection containers) for measurement of cortisol. On day 2, a low dose (0.5 mg) of dexamethasone is given by mouth every 6 hours for 48 hours.
For the high-dose overnight method, a baseline cortisol is measured on the morning of the test, then 8 mg of dexamethasone is given at 11 p.m. Blood is drawn at 8 a.m. for a cortisol measurement. For the standard high-dose test, urine is collected over 3 days (stored in 24-hour collection containers) for measurement of cortisol. On day 2, a high dose (2 mg) of dexamethasone is given by mouth every 6 hours for 48 hours.
What abnormal results mean:
If there is not a normal response on the low-dose test, abnormal secretion of cortisol is likely (Cushing's Syndrome). This could be a result of a cortisol-producing adrenal tumor, a pituitary tumor that produces ACTH, or a tumor in the body that inappropriately produces ACTH. The high-dose test can help distinguish a pituitary cause (Cushing's Disease) from the others.
Cushing's syndrome caused by adrenal tumor
- Low dose: no change
- High dose: no change
- Low dose: no change
- High dose: no change
- Low dose: no change
- High dose: normal suppression
Saturday, January 7, 2012
What is the main constituent of the hilar shadows seen radiographically?
The hilar shadows seen on radiography are caused mainly by branches of the pulmonary arteries, although the larger bronchi can frequently be recognized.
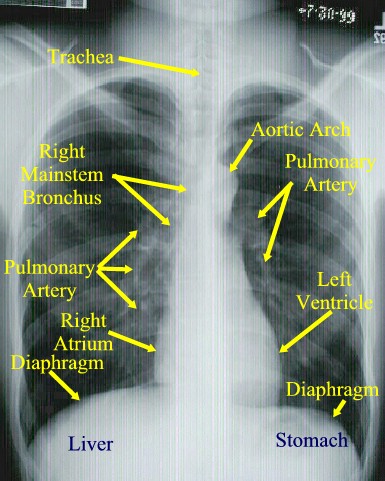
http://www.dartmouth.edu/~humananatomy/part_4/chapter_22.html
http://www.dartmouth.edu/~humananatomy/part_4/chapter_22.html
Saturday, December 31, 2011
Tuesday, December 27, 2011
Sotos syndrome
Sotos syndrome (cerebral gigantism) is a rare genetic disorder characterized by excessive physical growth during the first 2 to 3 years of life. The disorder may be accompanied by autism[1] mild mental retardation, delayed motor, cognitive, and social development, hypotonia (low muscle tone), and speech impairments. Children with Sotos syndrome tend to be large at birth and are often taller, heavier, and have larger heads (macrocephaly) than is normal for their age. Signs of the disorder, which vary among individuals, include a disportionately large and long head with a slightly protrusive forehead, large hands and feet, hypertelorism (an abnormally increased distance between the eyes), and downslanting eyes. Clumsiness, an awkward gait, and unusual aggressiveness or irritability may also occur. Although most cases of Sotos syndrome occur sporadically, familial cases have also been reported. It is similar to Weaver syndrome.
http://en.wikipedia.org/wiki/Sotos_syndrome

http://en.wikipedia.org/wiki/Sotos_syndrome
Prader–Willi syndrome
Prader–Willi syndrome (abbreviated PWS) is a rare genetic disorder in which seven genes (or some subset thereof) on chromosome 15 (q 11–13) are deleted or unexpressed (chromosome 15q partial deletion) on the paternal chromosome. It was first described in1956 by Andrea Prader (1919–2001), Heinrich Willi (1900–1971), Alexis Labhart (1916), Andrew Ziegler, and Guido Fanconi of Switzerland.[2] The incidence of PWS is between 1 in 25,000 and 1 in 10,000 live births. The paternal origin of the genetic material that is affected in the syndrome is important because the particular region of chromosome 15 involved is subject to parent of origin imprinting, meaning that for a number of genes in this region only one copy of the gene is expressed while the other is silenced through imprinting. For the genes affected in PWS, it is the paternal copy that is usually expressed, while the maternal copy is silenced. This means that while most people have a single working copy of these genes, people with PWS have no working copy. PWS has the sister syndrome Angelman syndrome in which maternally derived genetic material is affected in the same genetic region.
http://en.wikipedia.org/wiki/Prader%E2%80%93Willi_syndrome
,_de_Juan_Carre%C3%B1o_de_Miranda..jpg)
http://en.wikipedia.org/wiki/Prader%E2%80%93Willi_syndrome
Noonan syndrome
Noonan syndrome is a disease passed down through families (inherited) that causes abnormal development in many parts of the body. It used to be called Turner-like syndrome.
Causes, incidence, and risk factors
Noonan syndrome is linked to defects in several genes. Problems with the genes cause certain proteins involved in growth and development to become overactive.
Noonan syndrome is an autosomal dominant condition. This means only one parent has to pass down the faulty gene for the baby to have the syndrome. However, some cases may not be inherited.

Subscribe to:
Posts (Atom)